MASSパッケージのサンプルデータセットを巡回中。大文字優先のABC順。前回ビーバー1だったので、まだビーバー2が残ってますが昔のデータに別な処理をするだけなのはトキメかないので先に進みました。今回は biopsy です。乳がん患者様の生検データみたいです。良性か悪性か判断するためにデータを処理する基本的処理?
※「データのお砂場」投稿順Indexはこちら
Biopsy Data on Breast Cancer Patients
腫瘍が、良性(benign)なんだか悪性(malignant)なんだか判定するための基準を作るためのサンプル・データ・セットみたいです。解説ページが以下に。
https://stat.ethz.ch/R-manual/R-devel/library/MASS/html/biopsy.html
データ・セットには合計11変数(列)が含まれており、以下のようです。
-
- class列、目的変数、腫瘍が悪性か良性か
- V1~V9列、説明変数、生検対象の特徴
- ID列は各データのID
V1~V9列について、素人老人が整理すると以下のようです。
-
- V1 clump thickness. 細胞塊の厚さ
- V2 uniformity of cell size. 細胞サイズの均一性
- V3 uniformity of cell shape. 細胞形状の均一性
- V4 marginal adhesion. 辺縁癒着
- V5 single epithelial cell size. 単一の上皮細胞の大きさ
- V6 bare nuclei 裸核
- V7 bland chromatin. おとなしいクロマチン(クロマチンが均一)
- V8 normal nucleoli. 正常な核小体
- V9 mitoses. 有糸分裂
例えばV1の「細胞塊の厚さ」は、健康な細胞は通常均一で層も薄いらしいですが、がん細胞は厚いのだそう。しかし「正常な核小体」などといわれても素人老人には何のことやらサッパリです。まあプロが顕微鏡を覗き込むと分かるのでしょう。すべての説明変数は1から10の数値で表されてます。
先ずは生データ
生データをロードして、眺めたところが以下に。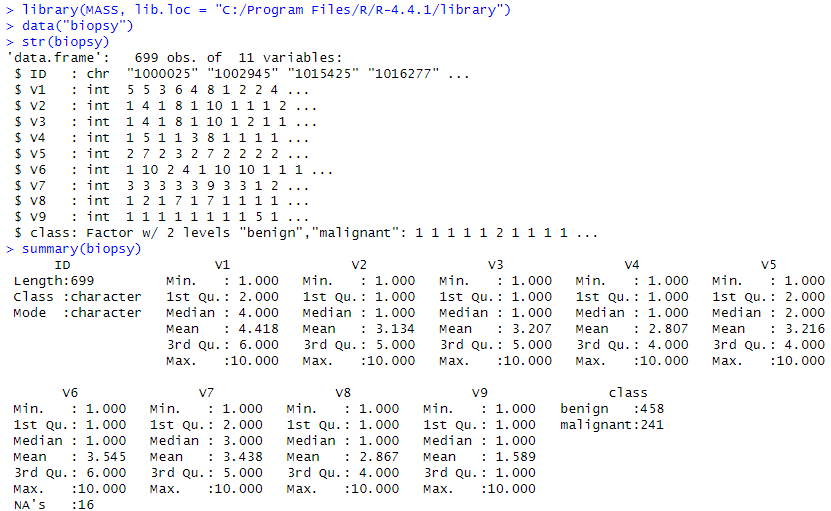
まあね、生データをロードしてみたものの、ここからどしたら良いの?
困ったときの生成AI頼み、いつものようにGoogleの生成AI、Gemini 2.5 Flash様にお聞きしながら以下作業してみました。
まずは下準備だと
上の summaryをみると気づくのですが、V6変数のところに 「NA’s : 16」という表示があります。欠損値が含まれているみたい。以下のようにしても欠損値を数えることができます。
まあ、V6だけみたいね。とりあえず欠損値のあるレコードは御引取願うことにして、サブセットなデータベース biopsy.w を生成
nrow(biopsy) biopsy.w <- na.omit(biopsy) nrow(biopsy.w)
結果はこんな感じ。元699件あったレコードが683件と16件減りました。ここは予定通りね。
つづいて、Gemini様の御教えは、目的変数が文字列なので、ファクタ化してしまえ、ついでに処理に使わないIDフィールドは落としてしまえ、とのこと。素直にやってみます。
biopsy.w$class <- factor(biopsy.w$class) biopsy.w <- subset(biopsy.w, select = -ID) str(biopsy.w)
この結果が以下に。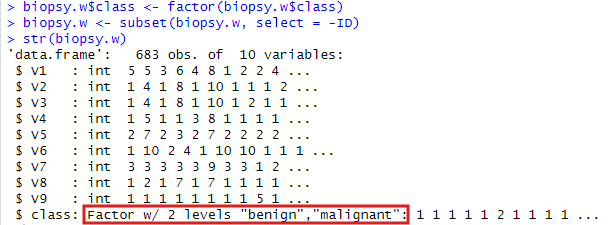
各説明変数毎に目的変数との関わりを観察
まずは、各説明変数毎に箱ヒゲ図を描いてみました。頭を使わない力業の処理が以下に。
layout(t(matrix(1:9, nr=3)))
boxplot(V1 ~ class, data = biopsy.w, main = "V1 (Clump Thickness)", xlab = "Class", ylab = "V1 Score (1-10)", col = c("skyblue", "salmon"))
boxplot(V2 ~ class, data = biopsy.w, main = "V2 (uniformity of cell size)", xlab = "Class", ylab = "V2 Score (1-10)", col = c("skyblue", "salmon"))
boxplot(V3 ~ class, data = biopsy.w, main = "V3 (uniformity of cell shape)", xlab = "Class", ylab = "V3 Score (1-10)", col = c("skyblue", "salmon"))
boxplot(V4 ~ class, data = biopsy.w, main = "V4 (marginal adhesion)", xlab = "Class", ylab = "V4 Score (1-10)", col = c("skyblue", "salmon"))
boxplot(V5 ~ class, data = biopsy.w, main = "V5 (single epithelial cell size)", xlab = "Class", ylab = "V5 Score (1-10)", col = c("skyblue", "salmon"))
boxplot(V6 ~ class, data = biopsy.w, main = "V6 (bare nuclei)", xlab = "Class", ylab = "V6 Score (1-10)", col = c("skyblue", "salmon"))
boxplot(V7 ~ class, data = biopsy.w, main = "V7 (bland chromatin)", xlab = "Class", ylab = "V7 Score (1-10)", col = c("skyblue", "salmon"))
boxplot(V8 ~ class, data = biopsy.w, main = "V8 (normal nucleoli)", xlab = "Class", ylab = "V8 Score (1-10)", col = c("skyblue", "salmon"))
boxplot(V9 ~ class, data = biopsy.w, main = "V9 (mitoses)", xlab = "Class", ylab = "V9 Score (1-10)", col = c("skyblue", "salmon"))
上記の処理結果のプロットが以下です。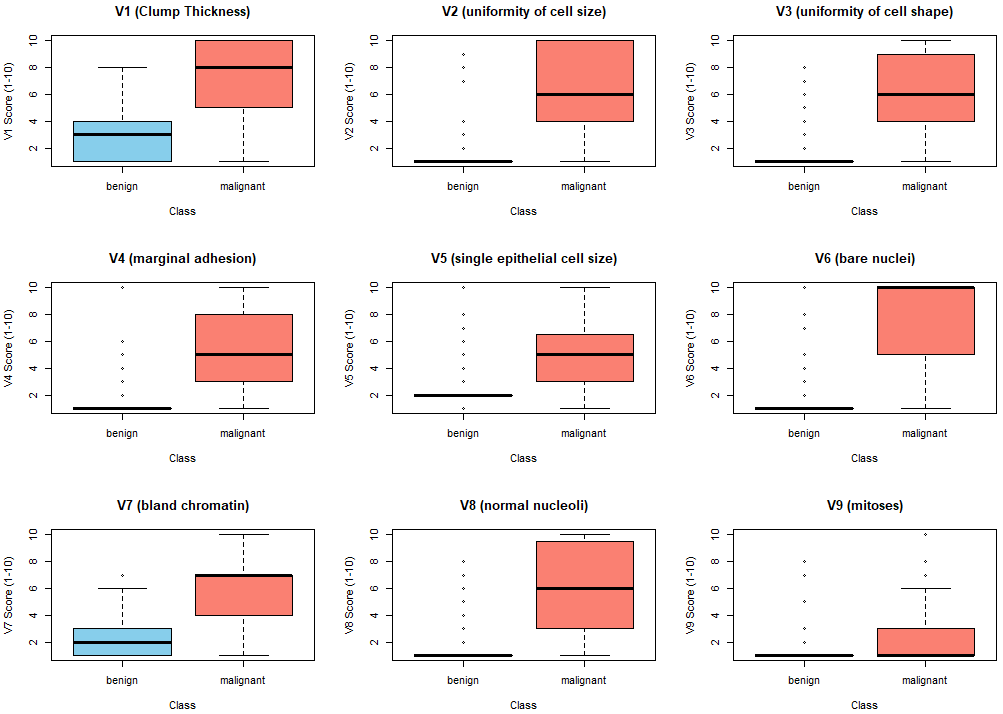
だいたいどの説明変数も、数字が大きい方が悪性、小さければ良性っぽいという傾向みたい。ただ、結構例外もあるのね。
線形判別分析: LDA
Gemini様の御教えを読んでいると、このサンプルデータセットに適用するべき処理は、MASSパッケージ所蔵の関数 lda (線形判別分析、Linear Discriminant Analysis)であるようです。どうもこの手のデータの処理の定番?みたい。ホントか?
処理はこんな感じ。
lda_model <- lda(class ~ V1 + V2 + V3 + V4 + V5 + V6 + V7 + V8 + V9, data = biopsy.w) print(lda_model)
結果が以下に。色付文字は素人老人が書き加えたもの。
bare nucleiって何?、裸核? 核膜のナイ細胞核?なんだそれ。まあ、なんだかんだで全9個の変数の中でも、重要度が高いヤツもあれば、あまり効かないヤツもいるということは分かった。「効くヤツ」だけにしてしまう?